
1、INTRODUCTION
Venous leg ulcers are chronic wounds affecting approximately 1% of Americans, and are more prevalent in the elderly population (aged 65 years and older).1 They are difficult to treat and can cause significant morbidity in patients who are not able to modify the causative factors for the lesions or who are not surgical candidates. VLUs are recurrent in 72% of patients diagnosed and have the worst healing prognosis of all leg ulcers.2 Typically, VLU wounds remain open for more than a year highlighting the need for prolonged management and more effective treatment strategies.3 VLUs are a late response of venous hypertension in the lower extremity. This hypertension can be caused by inadequate muscle pump function, incompetent valves or venous thrombosis. When venous pressure is elevated, it is difficult to achieve arterial perfusion and thus oxygen delivery, which can both cause the ulcer and retard wound healing. Due to the resultant poor arterial circulation in the area of these ulcers, many of the cellular and proteinaceous elements needed for wound healing are not able to access the wound bed.4 Interestingly, even when venous hypertension is mitigated, either surgically or by modifications in comorbidities, VLUs may persist or recur. This suggests that more research is needed to fully understand the pathophysiology of these ulcers.
Recently, the interplay between the microbiome and the wound environment has been shown to play a role in both chronic wound development5,6 and wound healing.7,8 The precise impact of the microbiome on wounds can be tied to microbial diversity of the host skin, where microbial composition varies depending on moisture content of the skin as well as dermal versus epidermal colonization. 9-11 Highly represented epidermal skin bacterial phyla include Actinobacteria, Firmicutes, Proteobacteria, and Bacteroidetes, whereas subepidermal compartments typically contain higher proportions of Proteobacteria and Actinobacteria,9-11 Interestingly, unlike the gut and other microbiome systems in which increased diversity of host commensal organisms correlates with improved outcomes in dysbiotic conditions, the role of bacterial diversity in chronic wounds is less straight-forward. Studies examining diabetic ulcers, VLUs, and other chronic wounds show increases or decreases in the diversity of skin bacteria in wounds accompanying positive healing outcomes.12-14 In fact, treatments that disrupt the wound microbiome and cause either increases or decreases in diversity may result in faster healing. 7,8 The variability of the wound microbiome suggests that successful treatment is in part dependent on the individual's microbial ecology and treatment regimen.
Chronic wounds are colonized by a diverse array of bacterial genera, and one mechanism that is hypothesized to contribute to chronicity is the presence of bacterial biofilms.15 In ulcer environments, commensal skin bacteria may contribute to the formation of biofilms leading to persistent infection, particularly when host immune function is impaired.16 One study found that the most abundant genera in diabetic foot ulcers included Staphylococcus and Corynebacterium. 17 Also, anaerobes are found in high abundance due to low oxygen presence in the wound yet are not generally detectable by traditional clinical culturing methods.18 If a biofilm is detectable in VLUs, usually by biopsy, the presence of Staphylococcus aureus and Pseudomonas accounts for a large percentage of the bacteria in the biofilm, and these bacteria are believed to initiate biofilm formation.19 Biofilms in wounds are mostly polymicrobial; however, monomicrobial biofilms are seen as well in a much smaller percentage. Finally, wound depth can determine the presence or absence of particular bacteria.20 Various wound types sampled in different anatomical locations across patients showed a predominance of Staphylococcus residing in superficial wounds, while Pseudomonas persisted in deeper wound layers.6
Treatment of chronic VLUs consists of topical ointments, creams, foams, hydrogels, and other dressing products developed to promote a positive wound healing environment. Chlorhexidine-based antiseptic regimens of the skin have been shown to promote a decrease in bacterial burden and infection in surgical wounds.21 The effects of antiseptics on resident skin epithelial inhabitants have been characterized as short-term with an increase in predominant genera and loss of less abundant genera; effects are highly personalized to skin site following antiseptic treatment.22 Treatment of VLUs follows a regimen of topical antiseptic to the site of the wound followed by application of a secondary dressing that allows for compression. These dressings are changed at intervals of 7–10 days. At dressing change, the wounds are assessed, debrided, and then cleansed or washed with an antimicrobial solution before new dressings are applied. Systemic antimicrobial treatments may be used in certain circumstances but have little efficacy for promoting chronic wound healing due to antibioticresistant bacteria present in large numbers in biofilms.3
Although modulation of the wound microbiome is becoming a promising modality for therapy in wound treatment, a comprehensive understanding of microbiome dynamics before and after therapy in VLUs is currently lacking. Therefore, in this study, we undertook a survey of 11 patient microbiomes by performing 16S amplicon sequencing and in vitro culture using a selection of bacteria-specific media on skin swabs isolated from VLUs before and after wound washing. Bacterial composition of wound samples before and after wash were quantified and compared to determine effects of washing on bacterial communities. Results from this study may have implications for the success of therapeutic strategies in treating this disease.
2、METHODS
2.1 Sample collection
Eleven patients with persistent VLUs established for >1 year were included in this study. All patients received the same primary dressing and compression as well as dressing change interval. Wound cleansing was performed with chlorhexidine gluconate and hypochlorous acid solutions for all patients. Information regarding wound debridement was not collected as part of this study and may or may not have been performed as part of standard of care treatment for this patient cohort. Swabs of the wound were collected immediately before and immediately after washing. Swabs were obtained using the Levine technique23 by rotating the swab 360 degrees in a 1 cm square area for 5 s using gentle pressure to release tissue exudate, and samples were placed in cryovials containing 1.5 ml of tryptic soy broth or placed in a cryovial and covered in AllProtect Tissue Preservation Reagent (Qiagen). Tips were broken off into the tubes. The swab tips in broth were processed within approximately 30 min of collection in the laboratory for plating and assessment of bacterial growth on selective media. The swabs in AllProtect Reagent to be used for molecular work were stored at -80° C. Wounds were washed and then swabs were collected immediately following wash using the same procedure as pre-wash.
2.2 Wound characterization and treatment
All wounds were located overlying the medial malleolus. Wounds were treated with a primary dressing that contained ACTICOAT (Smith Nephew) followed by Drawtex (Urgo). A secondary dressing for compression was the 3M™ Coban™ 2 Layer Compression System. This dressing regimen remained in place for a week.
2.3 Bacterial growth on selective growth plates
Swabs from before-wash and after-wash were thoroughly vortexed in media. The tip of the swab was then removed from the media. Media was serially diluted, and plated on tryptic soy agar, MacConkey agar, Streptococcus selective agar, and mannitol salt agar growth selective plates and allowed to grow 24 h at 37°C. Samples were plated in duplicate. Bacterial growth was quantified as present or absent for growth following 24 h.
2.4 DNA extraction and 16S rRNA sequencing
Genomic DNA was extracted from 22 swab samples using the DNeasy PowerWater Kit (Qiagen) and normalized on an Eppendorf epMotion 5075 Liquid Handling Workstation to 0.2 ng/ml. Primers were designed to target the 16S V3 and V4 regions (forward primer: 50 -TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNG GCWGCAG-30 , reverse primer: 5'-GTCTCGTGGGCTCGGAGATGTG TATAAGAGACAGGACTACHVGGGTATCTAATCC-30 ). The targeted regions were amplified via PCR, purified, attached to Illumina sequencing adapters using the Nextera XT Index Kit Set A, and purified again before library pooling. After quality control, final libraries were sequenced on the Illumina MiSeq using v3 reagents with a 600 bp kit (paired 300-bp reads) in a single 65-h run.
2.5 16S amplicon data processing and bacterial identification
Reads underwent quality filtering using Trimmomatic (version 0.35), where adapter sequences were eliminated, and reads were cut at points of low quality using a sliding window of 4 and a minimum PHRED score of 20. After read quality control, paired-end reads were joined using QIIME's join_paired_ends.py script with default settings. Unjoined reads were discarded and assembled reads were assigned to samples from barcodes using QIIME's split_libraries.py.24 OTUs were identified by open reference OTU picking using the GreenGenes25 13_5 97% database and QIIME's pick_open_reference_otus.py script. For downstream analysis, OTUs present >1% in at least one sample were included.
2.6 Diversity analysis
All diversity analyses utilized the percent abundance table where OTUs present >1% in at least one sample were included. The percent abundance table was analysed using the Bray–Curtis dissimilarity metric as a measure of β-diversity between before- and after-wash samples within a patient, before-wash samples across patients, and after-wash samples across patients. Cluster analysis of before-wash samples was performed using hierarchical clustering of the Bray–Curtis dissimilarity metric. The optimal cluster number of 4 was determined by calculating the consistency of clusters with the silhouette technique, a quantification of how similar an object is to its assigned cluster compared to neighbouring clusters. The maximum average silhouette is considered the appropriate number of clusters. Clustering arrangements 1–10 were tested using the factoextra R package (Figure S1B). Statistical significance of hierarchical clusters was tested using pvclust boostrap analysis of clustering with threshold of au >90% significance.26 To quantify α-diversity, Shannon diversity (H) was calculated for all samples using H = –Σpi▪ln(pi ), where pi represents the normalized population fraction of species i. The number of unique OTUs represented in a sample was also used as a metric of α-diversity. To test whether α-diversity changed between beforewash and after-wash samples across all patients, a paired Student's t-test was used on Shannon diversity quantifications and total unique OTUs on before and after samples.
2.7 Data availability
Raw sequencing data are available from the Short Read Archive (SRA) with accession number PRJNA704944.
3、RESULTS
3.1 Characterization of patient venous stasis ulcer microbiome
We investigated the wound bacterial microbiome ecology of 11 patients with established VLUs by collecting skin swabs from wounds following removal of dressings and compression bandages. Swabs were collected prior to and after washing with either a hypochlorous acid- or a chlorohexidine-based soap solution. We performed 16S amplicon sequencing on these before-wash and afterwash swab samples, obtaining libraries with an average sequencing depth of approximately 46,000 reads per sample. From these data, we were able to identify 44 different genera present in any sample at >1% relative abundance, a commonly employed threshold to distinguish low abundance genera from common or high abundance genera clinically27 (Tables 1 and S1).
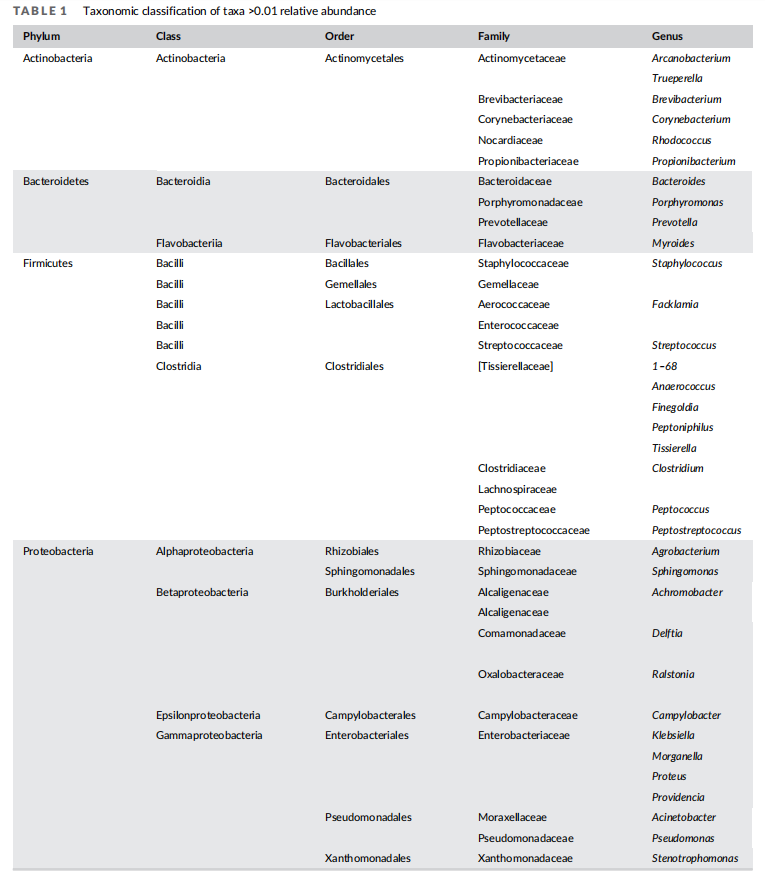
Previous studies have shown VLU microbiomes comprised a diverse bacterial population that varies depending on patient. To examine diversity of the bacterial populations in VLUs in this patient cohort, we quantified the relative abundance of bacterial genera in each patient before wash. We found the top 10 most ubiquitous genera to contain mostly gram-negative bacteria with some representation of gram-positive bacteria. Organisms included aerobes, anaerobes, and facultative anaerobes (Table 2). The top five genera in order of most to least abundant were Proteus, Pseudomonas,Morganella, Providencia and Finegoldia. Of these, Morganella was unique to this patient population compared to several studies examining microbiome in VLU patients16,28 (Figure 1(A)).
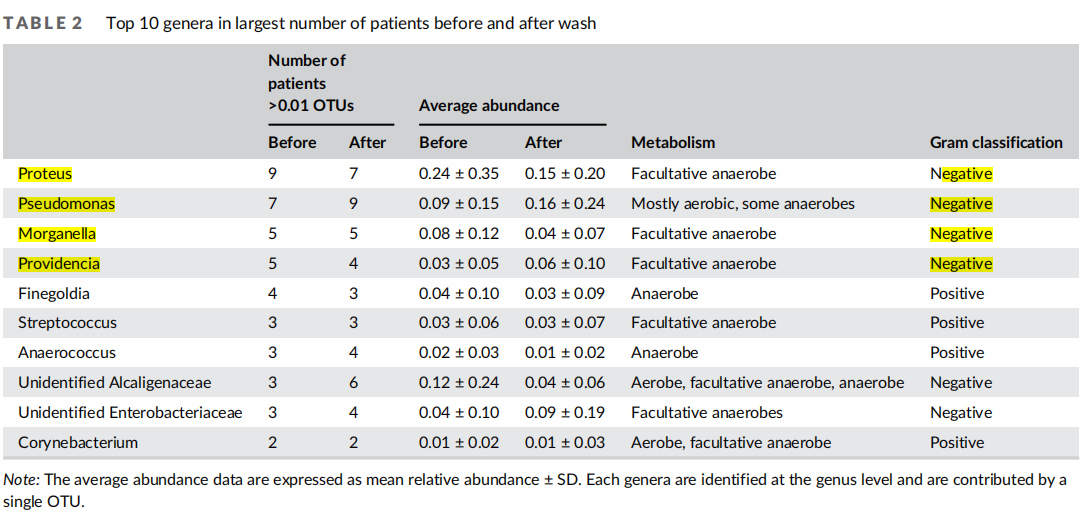
Cluster analysis based on the Bray-Curtis index of relative abundance of shared genera across patients revealed that patients formed four unsupervised clusters (Figure 1(B)) as determined by comparison of intra- and inter-cluster distances through silhouette analysis (Figure S1B). Significance of hierarchical clusters was determined using a multi-scale bootstrap resampling technique on all clusters (4 clusters = pval >90%, Figure S1C). Principal coordinate analysis of the Bray-Curtis distance indices also displayed four groups when the top three most variable axes were measured (total variance captured 60.8%) (Figure 1(C)). To determine patterns in community composition of each of the four clusters, we examined alpha diversity, a measure of community richness and evenness within clusters (Figure 1(D)). We found that cluster 3 had reduced diversity compared to other clusters. We next examined the number and composition of genera in each patient (Figure 1(E)). We found that each patient varied in the community members present in samples before wash, yet a few taxonomic features were consistent within each cluster (Table 3). Cluster 1 contained a single patient (P-42) predominantly colonized by four genera, the most abundant of these genera being an unidentified genus from the family Oxalobacteraceae. Cluster 2 patients (P-33, P-41, and P-43) were colonized mainly by an unidentified genus from Alcaligenaceae (18%–64%) and had 7–9 genera total. Cluster 3 patients (P-28, P-30, and P-40) were colonized by Proteus as the dominant genera (45%– 90%) and were colonized by fewer genera overall (2–4) than other patients in this cohort, suggesting reduced diversity of the microbiome in this patient subset. All Cluster 4 patients exhibited a large abundance of Morganella. These data suggest that there may be underlying characteristics in the wound environment promoting growth of particular genera in some patients in this cohort.
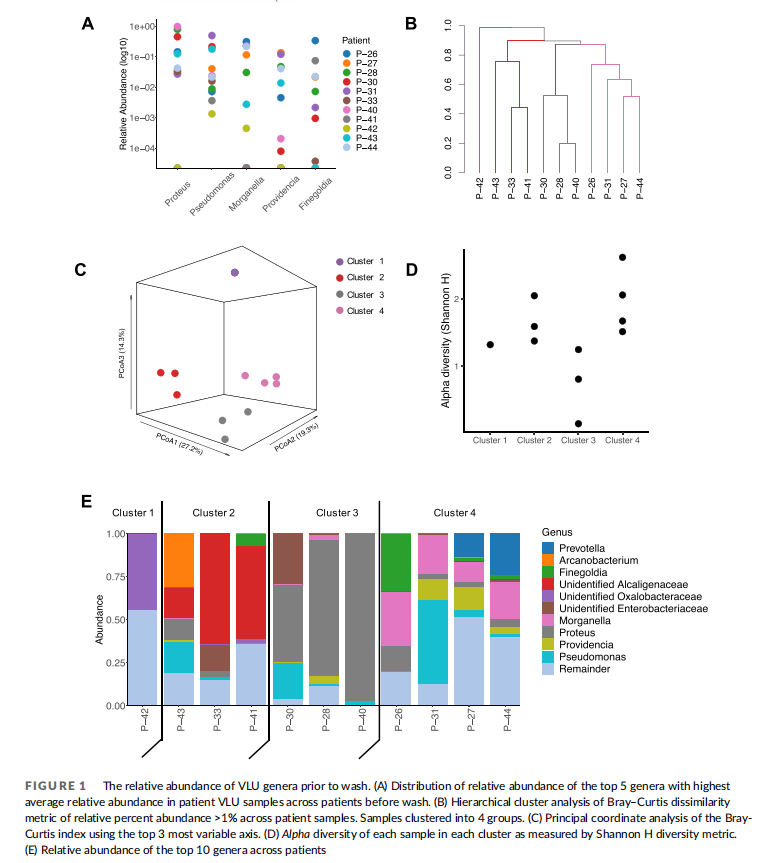
3.2 Washing venous stasis ulcers does not change diversity of the ulcer microbiome
Previous studies examining VLU microbiomes report contradictory results regarding microbiome diversity in cases of healed or unhealed wounds. In some cases, diversity is either unchanged or higher in wounds that did not heal, and in other cases, no correlation is found regarding length of time of wound healing or treatment with antibiotics. 14,28 To understand the effect of washing of VLUs on microbiome diversity, we quantified patient samples for metrics of microbiome diversity within sample before and after washing across this patient cohort (Figure 2(A), (B)). We first examined the number of genera >1% in each patient's set of samples before and after washing and compared whether there was effect of washing on species richness across the patient cohort (Figure 2(A)). We found no difference overall in species richness before and after washing (p-value 0.42, Student's paired t-test). We also examined the relative proportion of each species within a sample as a measure of diversity using the Shannon diversity index and compared samples before and after washing across the dataset. No statistical difference was detected across the dataset in evenness of the distribution of genera (p value 0.43, Student's paired t-test) (Figure 2(B)). Interestingly, when examining genera richness and genera diversity within individual patients, some notable differences in alpha diversity could be detected (Figure 2(C), (D)). However, additional data are required to confirm these observations. When quantifying number of genera in beforeand after-wash samples by patient (present at >1% relative abundance), we found 3/11 patients lost detected genera, 5/11 patients gained detected genera, and 3 patients experienced no change in detected genera (Figure 2(C)). Also, comparison of Shannon indices before and after washing for each patient revealed an approximately even split between patients with increased diversity and patients with decreased diversity following washing (Figure 2(D)). The lack of significant difference in alpha diversity metrics including richness and diversity suggests that washing does not affect diversity of patient microbiome samples in this cohort.
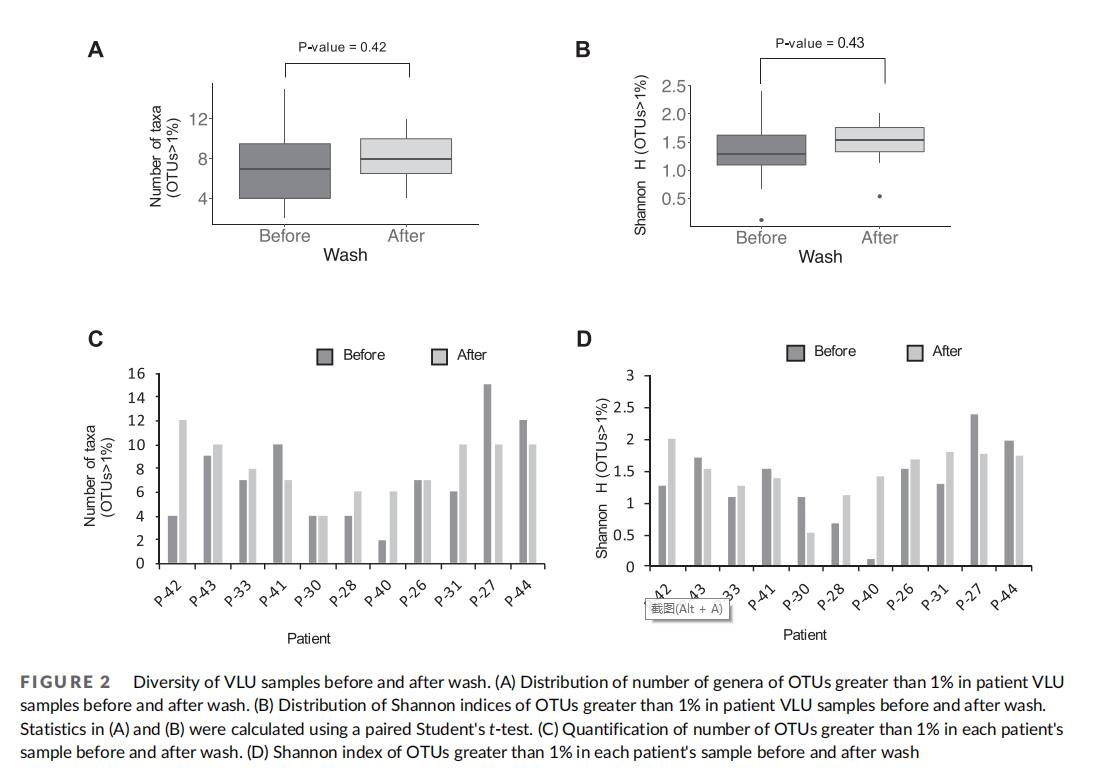
3.3 Modulation of community structure of venous stasis ulcers following washing
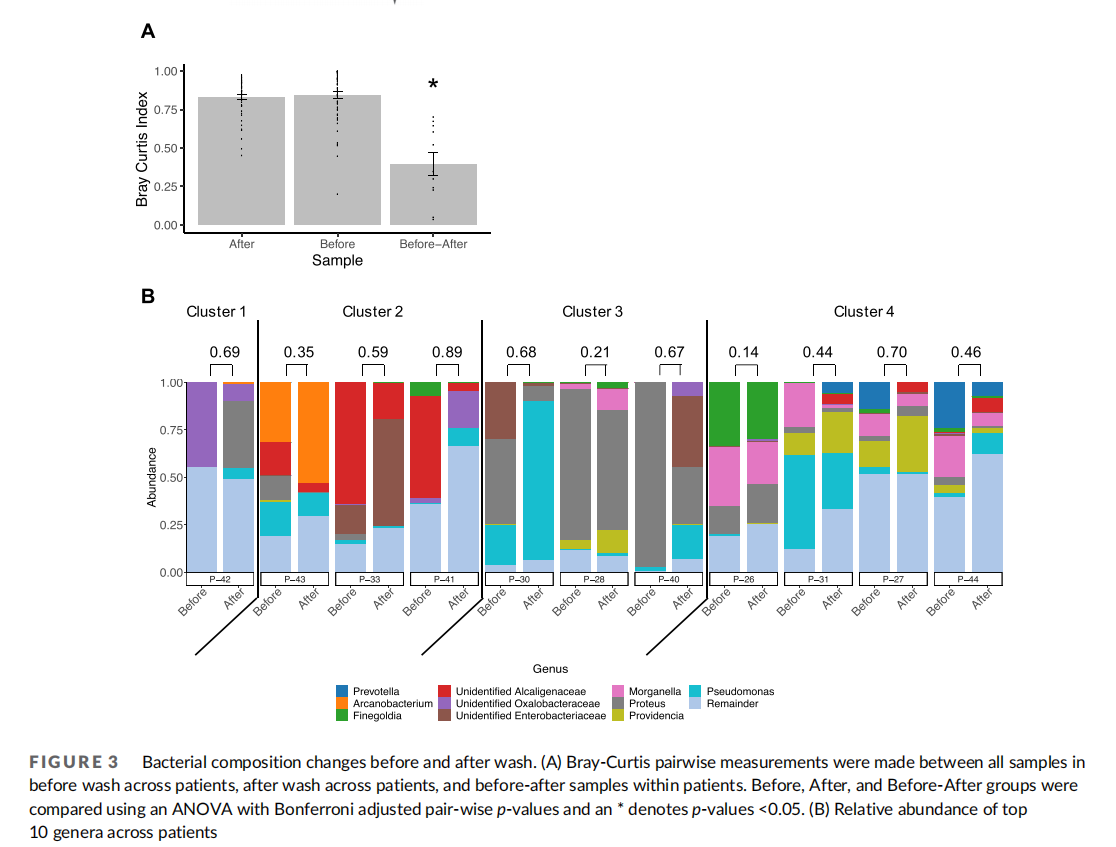
Though overall species richness and evenness were unchanged by washing in our patient population, specific community composition of wounds may be affected by the washing procedure itself, particularly if washing affects the ability of certain bacteria to adhere to the wound. Specific community structure of VLUs across samples in this patient cohort before wash revealed some similarities within clusters of patients' VLU microbiomes (Figure 1(B)–(D)). Therefore, we next examined changes in community structure associated with washing within each patient and each patient cluster. First, we assessed interpersonal variation of all samples before washing and after washing using Bray-Curtis index and found that samples had a high level of variation in community structure in both before and after samples across patients (Figure 3(A)). When comparing intrapersonal variation of samples before and after washing within each patient, community structure variation was reduced. This finding suggests that community structure is more similar within a patient than across patients, supporting the high diversity of VLU microbiomes. Previous studies surveying the healthy skin microbiome9,10 have similarly found that interpersonal variation is greater than intrapersonal variation.
To address whether particular bacteria were more or less affected by the washing procedure, we compared the relative abundance of each bacterial genera across patients before and after washing (Figure 3(B)). We found that no bacterial genera had increasing or decreasing trends following wound washing across all patients. However, particular patients exhibited increases or decreases in specific genera. Moreover, strikingly, the salient genera defining each cluster decreased after wash in all patients. Namely, Oxalobacteraceae decreased in P-42 defining Cluster 1, Alcaligenaceae decreased in all Cluster 2 patients, Proteus decreased in all Cluster 3 patients, and Morganella decreased in all Cluster 4 patients (Table 3). Interestingly, 2 of the 3 patients in Cluster 3 exhibiting the most pronounced decrease in Proteus following washing displayed concomitant increases in Pseudomonas (P-30, P-40), though in other clusters, increases in Proteus following washing did not correlate to a decrease in Pseudomonas (P-42). We also found no correlation in number of genera with Bray–Curtis dissimilarity measure (Figure 3(A)) before and after wash, suggesting that the degree of change in community structure following wash is not tied to the number of genera present before wash.

3.4 Culture-based analysis reveals decrease in abundance of viable bacteria
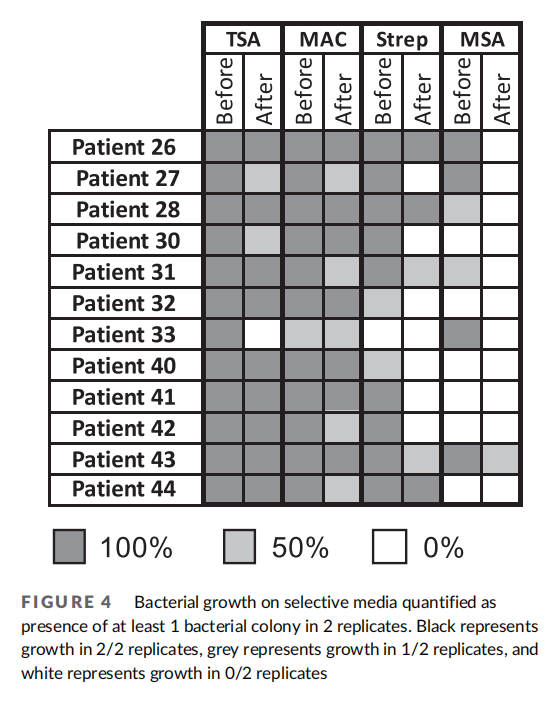
We qualitatively measured the presence or absence of bacteria using standard culturing methods as complimentary analysis to our 16S rRNA gene sequencing results (Figure 4). We found that samples before wash unanimously had bacterial growth on tryptic soy agar (TSA). Most samples excluding P-33 contained gram-negative bacteria in their pre-wash samples as seen by presence of bacteria on MacConkey growth media plates. All samples except P-33 showed growth on Streptococcus selective plates in prewash samples. Six of 11 samples showed growth on MSA plates indicative of Staphylococcus or Micrococcus. After wash, one patient showed an absence of bacterial growth on TSA. Most patients' samples contained gramnegative bacteria. Interestingly, the majority of patients showed an overall loss in Streptococcus and Staphylococcus/Micrococcus species after washing. These results suggest specific bacteria genera are affected by the washing process as several genera were newly detected or lost due to washing.
4、DISCUSSION
Our 16S sequencing classification of bacterial genera in VLU wounds illustrates that microbiome differences within and across patients before and after washing are diverse and individual to the patient. We identified that patients' microbiomes in wounds prior to washing ranged from being predominantly characterized by a few genera to many genera and that relative abundances of genera were highly variable across patients. Trends in the number and distribution of genera within patients before and after washing were variable, with both increases and decreases in diversity. Thus, no consistent trend in diversity was observed. Though the number of patients in this study was relatively small, we were able to find trends in VLU microbiome community structure within small clusters of patients prior to washing and a reduction in predominant genera within these groups of patients. Although washing did not consistently affect overall diversity of bacterial populations across patients, individual genera in certain patients displayed increases or decreases in abundance following washing treatment. Most strikingly, the salient genus within each cluster consistently decreased in relative abundance following washing.
Chronic wound infections have been shown to be polymicrobial,yet biofilms can also be predominantly populated with one bacterial species. Interestingly, before washing of wounds in this patient cohort, an average of 7 bacterial genera, and as many as 15 genera, could be detected. However, two patients had wounds with a single genera colonizing at more than 50% relative abundance. The varying degree to which patient samples were polymicrobial agrees with previous research that found that most wounds are polymicrobial, but a small percentage of wounds were predominantly colonized by one or two species.20
Bacteria ranging from commensals to known pathogens have been shown to form biofilms in studies of VLU microbiomes. In our study, patient wounds with predominantly one genus were comprised of Proteus. Proteus, an opportunistic pathogen, has been shown to be an infectious agent in soft tissue infections, and was seen as a dominant genera colonizing VLUs in another study, though only in a few patient samples.28 In diabetic foot ulcers with monoculture wounds, Proteus mirablis was associated with limb loss. In the same study, Morganella, another predominant colonizer in this study, and known contributor to soft tissue infections, was also found associated with limb loss in diabetic foot ulcers.29 Members of the family Alcaligenacea were also highly represented in two patient samples. Genera within Alcaligenacea are gram-negative rods found in a variety of environments including the human body, and have shown conflicting results in regards to promoting or inhibiting wound healing. Interestingly, one study has implicated Alcaligenes faecalis as a causative agent in skin and soft tissue infections in a small number of patients with vascular disease. 30 In contrast, a more recent study examining the microbiome of diabetic foot ulcers found that Alcaligenes faecalis promoted wound healing in an in vitro skin model suggesting that host, environment, or organism factors determine wound disease outcome in relation to this organism. 31 We also found that a genera identified as Pseudomonas was highly represented in patient samples, particularly before wash. P. aeruginosa is the most common producer of single organism biofilms and is sometimes associated with poor prognosis for woundhealin. 20 However, Pseudomonas representatives have also been shown to be able to colonize wounds without delaying wound healing, suggesting that organisms in this genus may have a diverse role in the wound microbiome.32 The presence of Proteus, Alcaligenacea, and Pseudomonas in patients in this dataset as predominating colonizers before wash may be suggestive of wounds that have formed biofilms and therefore may be more recalcitrant to therapy.
Previous studies have also found anaerobic bacterial species to predominate in the wound.8 In our study, 8 of the top 10 taxa were from genera usually identified as anaerobes. Wounds can be well aerated at the surface with oxygen becoming less available to deeper levels of the wound. Unlike typical clinical culturing methods, the methodology used in this study, including wound sampling technique, namely the application of pressure to express wound exudate, and isolation of bacterial DNA followed by targeted sequencing of 16S rRNA genes, allowed for identification of anaerobes in wound samples. 18
The stability of the microbiome in chronic wounds over time is associated with poor wound healing.7 Topical antibiotics and antiseptics are more efficacious when they are able to change the dynamics of the wound microbiome.7,27 In this study, wounds were washed with chlorohexidine gluconate and hypocholorous acid, each of which is commonly used independently in wound cleansing.33 Chlorohexidine, when used in isolation, has been shown in a previous study to confound the effects of DNA sequencing through retention of DNA on the skin surface leading to an inability to detect changes in genera or patient microbiome diversity post-wash. The authors hypothesized that the inflammatory reaction induced by chlorohexidine could lead to disruption of the skin barrier surface and promote DNA retention; however, the molecular mechanism of DNA retention was not formally tested in this study.22 In contrast, although wound washing did not affect overall diversity in our study population, washing was able to destabilize the microbiomes of several patients, as shown by the poor correlation of before and after samples, as well as changes in the relative abundance of bacteria. We hypothesize that we could capture microbiome changes because chlorohexidine and hypochlorous acid were added in combination. The addition of hypochlorous acid, which can reduce the inflammatory response and disrupt the polysaccharide matrix in biofilms,34 may ameliorate the immune-related skin barrier defects as well as potentially reduce general adhesion of DNA to skin. We were unable to find another study that examined these two wash solutions in combination. Thus, further characterization of skin inflammatory response and DNA retention in this context is an interesting avenue for future research.
A known caveat of sequencing is its inability to distinguish live and dead bacteria; however, culturing techniques suffer from an inability to grow all bacterial genera present in a sample. Though these drawbacks in experimental techniques exist, our study highlights that we are able to show shifts in structure of bacterial genera due to washing, suggesting that these techniques can in fact highlight trends in microbial community dynamics, particularly in combination. Indeed, culturing with media that specifically enriched for Streptococcus and Staphylococcus/Micrococcus revealed a decrease of these genera after wash in most patient samples, complimenting the sequencing results that captured increases and decreases in genera that could not be cultured. Results from both sequencing and culturing suggest a disruption of specific genera after wash, which could potentially be beneficial to the healing process. The knowledge that standard of care therapies can achieve disruption of microbiome genera may inform the frequency at which dressings need to be changed and duration of wash in order to have the wounds appropriately cleansed. Future studies examining correlations between washing, microbiome dynamics, and patient healing time will be informative to understanding possible mechanisms by which wound washing may be beneficial to healing through modulation of the microbiome.
Microbiome dynamics with respect to wound treatments are an actively researched area, yet studies have mainly focused on temporal dynamics of chronic wounds with respect to healing time. Few studies have focused on the effects of treatment modalities, particularly standard of care wound therapies, on the microbiome.7,27 Our study examined a standard of care therapy, wound washing, prior to applying dressing to the wound, and its effects on wound microbiome dynamics, adding to the knowledge from existing studies examining chronic wound microbiomes and effects of standard of care therapies.22,36,37 Washing and wound debridement, or removal of dead tissue from the wound, is a common standard-of-care therapy with a proven track record of success in regards to improvements in healing outcomes.35,36,37 Verbanic et al. examined the immediate effects of wound debridement on the wound microbiome of patients and found no difference in diversity between the wound pre- and post- debridement. Like Verbanic et al., we found no global increase or decrease in overall diversity of bacteria with standard of care therapy. We did not record debridement as part of standard of care for this patient cohort; however, we recognize that there could potentially be differences in specific genera in patient microbiomes that may be due to debridement in conjunction with washing. Though diversity of microbiomes was unaffected by washing regimens, individuals in the cohort displayed large shifts in microbiome composition with poor correlation in relative abundances of bacteria before and after wash. Interestingly, wounds with shifting microbiomes may promote wound healing, particularly when a pathogenic bacteria is displaced from the wound niche.7 Washing, in this patient cohort, had varying effects and it will be interesting to understand the impact of washing and microbiome dynamics on wound healing in future studies.
ACKNOWLEDGMENT
This material is based upon work supported by, or in part by, the U.S. Army Research Laboratory and the U.S. Army Research Office under contract/grant number #W911NF-14-1-0490.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ORCID
Amanda W. Ernlund https://orcid.org/0000-0003-1113-4103 David K. Karig https://orcid.org/0000-0002-9508-6411
REFERENCES
1、Margolisa DJ, Bilker W, Santanna J, Baumgarten M. Venous leg ulcer: incidence and prevalence in the elderly. J Am Acad Dermatol. 2002; 46:381-386.
2. Nelzén O, Bergqvist D, Lindhagen A. Venous and non-venous leg ulcers: clinical history and appearance in a population study. Br J Surg. 1994;81:182-187.
3. Tuttle MS. Association between microbial bioburden and healing out comes in venous leg ulcers: a review of the evidence. Adv Wound Care. 2015;4:1-11.
4. Agale SV. Chronic leg ulcers: epidemiology, aetiopathogenesis, and management. Ulcers. 2013;2013:1-9. https://doi.org/10.1155/2013/413604.
5.Chehoud C, Rafail S, Tyldsley AS, Seykora JT, Lambris JD, Grice EA. Complement modulates the cutaneous microbiome and inflammatory milieu. Proc Natl Acad Sci. 2013;110:15061-15066.
6. Sanchez CJ, Mende K, Beckius ML. Biofilm formation by clinical isolates and the implications in chronic infections. BMC Infect Dis. 2013; 13:47.
7. Loesche M, Gardner SE, Kalan L, et al. Temporal stability in chronic wound microbiota is associated with poor healing. J Invest Dermatol. 2017;137:237-244.
8. Johnson TR, Gomez BI, McIntyre MK. The cutaneous microbiome and wounds: new molecular targets to promote wound healing. Int J Mol. Sci. 2018;19:2699.
9. Grice EA, Kong HH, Renaud G, et al. A diversity profile of the human skin microbiota. Genome Res. 2008;18:1043-1050.
10. Grice EA, Kong HH, Conlan S, et al. Topographical and temporal diversity of the human skin microbiome. Science NY. 2009;324:1190-1192.
11. Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431.
12. Gardiner M, Vicaretti M, Sparks J. A longitudinal study of the diabetic skin and wound microbiome. PeerJ. 2017;5:3543.
13. Grice EA, Snitkin ES, Yockey LJ, et al. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci U S A. 2010;107:14799-14804.
14. Tuttle MS, Mostow E, Mukherjee P, et al. Characterization of bacterial communities in venous insufficiency wounds by use of conventional culture and molecular diagnostic methods. J Clin Microbiol. 2011;49:3812-3819.
15. Omar A, Wright JB, Schultz G, Burrell R, Nadworny P. Microbial biofilms and chronic wounds. Microorganisms. 2017;5:9.
16. Wolcott RD, Gontcharova V, Sun Y, Dowd SE. Evaluation of the bacterial diversity among and within individual venous leg ulcers using bacterial tag-encoded FLX and titanium amplicon pyrosequencing and metagenomic approaches. BMC Microbiol. 2009;9:226.
17. Lavigne JP, Sotto A, Dunyach-Remy C, Lipsky BA. New molecular techniques to study the skin microbiota of diabetic foot ulcers. Adv Wound Care. 2015;4:38-49.
18. Drago F, Gariazzo L, Cioni M, Trave I, Parodi A. The microbiome and its relevance in complex wounds. Eur J Dermatol. 2019;29:6-13.
19. Gjødsbøl K, Christensen JJ, Karlsmark T, Jørgensen B, Klein BM, Krogfelt KA. Multiple bacterial species reside in chronic wounds: a longitudinal study. Int Wound J. 2006;3:225-231.
20. Wolcott RD, Hanson JD, Rees EJ, et al. Analysis of the chronic wound microbiota of 2,963 patients by 16S rDNA pyrosequencing. Wound Repair Regen. 2016;24:163-174.
21. Patrick S, McDowell A, Lee A. Antisepsis of the skin before spinal surgery with povidone iodine-alcohol followed by chlorhexidine gluconate-alcohol versus povidone iodine-alcohol applied twice for the prevention of contamination of the wound by bacteria. Bone Jt J. 2017;99B:1354-1365.
22. SanMiguel AJ, Meisel JS, Horwinski J, Zheng Q, Bradley CW, Grice EA. Antiseptic agents elicit short-term, personalized, and body site–specific shifts in resident skin bacterial communities. J Invest Dermatol. 2018;138:2234-2243.
23. Levine NS, Lindberg RB, Mason AD, Pruitt BA. The quantitative swab culture and smear: a quick, simple method for determining the number of viable aerobic bacteria on open wounds. J Trauma. 1976;16: 89-94.
24. Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010; 7:335-336.
25. DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimerachecked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72:5069-5072.
26. Suzuki R, Shimodaira H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics. 2006;22:1540-
27. Tipton CD, Mathew ME, Wolcott RA, Wolcott RD, Kingston T, Phillips CD. Temporal dynamics of relative abundances and bacterial succession in chronic wound communities. Wound Repair Regen. 2017;25:673-679.
28. Davies CE, Hill KE, Wilson MJ, et al. Use of 16S ribosomal DNA PCR and denaturing gradient gel electrophoresis for analysis of the microfloras of healing and nonhealing chronic venous leg ulcers. J Clin Microbiol. 2004;42:3549-3557.
29. Hinojosa CA, Boyer-Duck E, Anaya-Ayala JE, et al. Impact of the bacteriology of diabetic foot ulcers in limb loss. Wound Repair Regen. 2016;24:923-927.
30. Tena D, Fernandez C, Lago MR. Alcaligenes faecalis: an unusual cause of skin and soft tissue infection. Jpn J Infect Dis. 2015;68:128-130.
31. Kalan LR, Meisel JS, Loesche MA. Strain- and species-level variation in the microbiome of diabetic wounds is associated with clinical outcomes and therapeutic efficacy. Cell Host Microbe. 2019;25:641- 655.e5.
32. Lipsky BA, Armstrong DG, Citron DM, Tice AD, Morgenstern DE, Abramson MA. Ertapenem versus piperacillin/tazobactam for diabetic foot infections (SIDESTEP): prospective, randomised, controlled, double-blinded, multicentre trial. Lancet. 2005;366:1695-1703.
33. Atiyeh BS, Dibo SA, Hayek SN. Wound cleansing, topical antiseptics and wound healing. Int Wound J. 2009;6:420-430.
34. Gold MH, Andriessen A, Bhatia AC, et al. Topical stabilized hypochlorous acid: the future gold standard for wound care and scar management in dermatologic and plastic surgery procedures. J Cosmet Dermatol. 2020;19:270-277.
35. Verbanic S, Shen Y, Lee J, Deacon JM, Chen IA. Microbial predictors of healing and short-term effect of debridement on the microbiome of chronic wounds. npj Biofilms Microbiomes. 2020;6:1-11.
36. Kalan L, Zhou M, Labbie M, Willing B. Measuring the microbiome of chronic wounds with use of a topical antimicrobial dressing – a feasibility study. PLoS One. 2017;12:e0187728.
37. Han G, Ceilley R. Chronic wound healing: a review of current management and treatments. Adv Ther. 2017;34:599-610.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
How to cite this article: Ernlund AW, Moffatt LT, Timm CM,et al. Examining the effect of wound cleansing on the microbiome of venous stasis ulcers. Wound Rep Reg. 2021;1–11. https://doi.org/10.1111/wrr.12926
This article is excerpted from the《Wound Repair and Regeneration》by Wound World.

